
- high-resolution terminology - matching measurements at high-resolution
MitoPedia: Ergodynamics
The MitoPedia terminology is developed continuously in the spirit of Gentle Science.
- What is ergodynamics?
- According to Ilya Prigogine [1], the importance of the thermodynamics of irreversible processes (TIP) can be gauged from its unique ability to assign general validity of phenomenological laws and discriminate these from special microscopic assumptions on molecular interactions. For this grand mission and promise to be fulfilled, the fundamental assumptions on the linear laws of TIP — implying linear relations between the generalized flows and forces called the phenomenological relations [2] — must primarily be shown to be generally valid.
- A critical isomorphic analysis provides the proof that the assumptions on the phenomenological relations are flawed by a confusion between isomorphic forces and pressures, rendering the variability of phenomenological coefficients L non-linearly dependent on the force under simple experimental conditions [3]. This shatters the foundation of the Onsager reciprocity relationships [2].
- The basis of the thermodynamics of irreversible processes, therefore, must be re-built on an entirely different paradigm deduced from the dynamic behavior of ergodic systems, where fluxes are linear functions of isomorphic pressures but not isomorphic forces [3]. To emphasize this radical paradigm shift, it is warranted to replace the designation of non-equilibrium thermodynamics or TIP by the term ergodynamics. In the footsteps of the thermodynamics of irreversible processes, the mission of ergodynamics is the revelation of relations of general validity.
- Prigogine I (1967) Introduction to thermodynamics of irreversible processes. Interscience New York, 3rd ed:147pp. - »Bioblast link«
- Onsager L (1931) Reciprocal relations in irreversible processes. I. Phys Rev 37:405-26. - »Bioblast link«
- Gnaiger E (2020) Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. 5th ed. Bioenerg Commun 2020.2:112 pp. https://doi.org/10.26124/bec:2020-0002
- The basis of the thermodynamics of irreversible processes, therefore, must be re-built on an entirely different paradigm deduced from the dynamic behavior of ergodic systems, where fluxes are linear functions of isomorphic pressures but not isomorphic forces [3]. To emphasize this radical paradigm shift, it is warranted to replace the designation of non-equilibrium thermodynamics or TIP by the term ergodynamics. In the footsteps of the thermodynamics of irreversible processes, the mission of ergodynamics is the revelation of relations of general validity.
- New in MitoPedia: Free activity
| Term | Abbreviation | Description | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acceleration | a, g [m·s-2] | Acceleration, a, is the change of velocity over time [m·s-2].
a = dv/dtThe symbol g is used for acceleration of free fall. The standard acceleration of free fall is defined as gn = 9.80665 [m·s-2]. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Activity | a | The activity (relative activity) is a dimensionless quantity related to the concentration or partial pressure of a dissolved substance. The activity of a dissolved substance B equals the concentration, cB [mol·L-1], at high dilution divided by the unit concentration, c° = 1 mol·L-1:
aB = cB/c° This simple relationship applies frequently to substances at high dilutions <10 mmol·L-1 (<10 mol·m-3). In general, the concentration of a solute has to be corrected for the activity coefficient (concentration basis), γB, aB = γB·cB/c° At high dilution, γB = 1. In general, the relative activity is defined by the chemical potential, µB aB = exp[(µB-µ°)/RT] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Advancement | dtrξ [MU] | In an isomorphic analysis, any form of flow is the advancement of a process per unit of time, expressed in a specific motive unit [MU∙s-1], e.g., ampere for electric flow or current, Iel = delξ/dt [A≡C∙s-1], watt for thermal or heat flow, Ith = dthξ/dt [W≡J∙s-1], and for chemical flow of reaction, Ir = drξ/dt, the unit is [mol∙s-1] (extent of reaction per time). The corresponding motive forces are the partial exergy (Gibbs energy) changes per advancement [J∙MU-1], expressed in volt for electric force, ΔelF = ∂G/∂elξ [V≡J∙C-1], dimensionless for thermal force, ΔthF = ∂G/∂thξ [J∙J-1], and for chemical force, ΔrF = ∂G/∂rξ, the unit is [J∙mol-1], which deserves a specific acronym [Jol] comparable to volt [V]. For chemical processes of reaction (spontaneous from high-potential substrates to low-potential products) and compartmental diffusion (spontaneous from a high-potential compartment to a low-potential compartment), the advancement is the amount of motive substance that has undergone a compartmental transformation [mol]. The concept was originally introduced by De Donder [1]. Central to the concept of advancement is the stoichiometric number, νi, associated with each motive component i (transformant [2]).
In a chemical reaction r the motive entity is the stoichiometric amount of reactant, drni, with stoichiometric number νi. The advancement of the chemical reaction, drξ [mol], is defined as, drξ = drni·νi-1 The flow of the chemical reaction, Ir [mol·s-1], is advancement per time, Ir = drξ·dt-1 This concept of advancement is extended to compartmental diffusion and the advancement of charged particles [3], and to any discontinuous transformation in compartmental systems [2], | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Advancement per volume | dtrY [MU∙L-1] | Advancement per volume or volume-specific advancement, dtrY, is related to advancement of a transformation, dtrY = dtrξ∙V-1 [MU∙L-1]. Compare dtrY with the amount of substance j per volume, cj (concentration), related to amount, cj = nj∙V-1 [mol∙V-1]. Advancement per volume is particularly introduced for chemical reactions, drY, and has the dimension of concentration (amount per volume [mol∙L-1]). In an open system at steady-state, however, the concentration does not change as the reaction advances. Only in closed systems and isolated systems, specific advancement equals the change in concentration divided by the stoichiometric number, drY = dcj/νj (closed system) drY = drcj/νj (general) With a focus on internal transformations (i; specifically: chemical reactions, r), dcj is replaced by the partial change of concentration, drcj (a transformation variable or process variable). drcj contributes to the total change of concentration, dcj (a system variable or variable of state). In open systems at steady-state, drcj is compensated by external processes, decj = -drcj, exerting an effect on the total concentration change of substance j, dcj = drcj + decj = 0 (steady state) dcj = drcj + decj (general) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Affinity of reaction | A [J·mol-1] | The concept of affinity and hence chemical force is deeply rooted in the notion of attraction (and repulsion) of alchemy, which was the foundation of chemistry originally, but diverted away from laboratory experiments towards occult secret societies [1].** Newton's extensive experimental alchemical work and his substantial written track record on alchemy (which he did not publish) is seen today as a key inspiration for his development of the concept of the gravitational force [2-4]. This marks a transition of the meaning of affinity, from the descriptive 'adjacent' (proximity) to the causative 'attractive' (force) [5]. Correspondingly, Lavoisier (1790) equates affinity and force [6]: “... the degree of force or affinity with which the acid adheres to the base” [5]. By discussing the influence of electricity and gravity on chemical affinity, Liebig (1844) considers affinity as a force [7]. This leads to Guldberg and Waage's mass action ratio ('Studies concerning affinity', 1864; see [5]), the free energy and chemical affinity of Helmholtz (1882 [8]), and chemical thermodynamics of irreversible processes [9], where flux-force relations are center stage [10].
According to the IUPAC definition, the affinity of reaction, A [J·mol-1], equals the negative molar Gibbs energy of reaction [11], which is the negative Gibbs force of reaction (derivative of Gibbs energy per advancement of reaction [12]): -A = ΔrF = ∂G/∂rξThe historical account of affinity is summarized by concluding, that today affinity of reaction should be considered as an isomorphic motive force and be generalized as such. This will help to (1) avoid confusing reversals of sign conventions (repulsion = negative attraction; pull = negative push), (2) unify symbols across classical and nonequilibrium thermodynamics [12,13], and thus (3) facilitate interdisciplinary communication by freeing ourselves from the alchemical, arcane scientific nomenclature. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Amount of substance | n [mol] | The amount of substance n is a base physical quantity, and the corresponding SI unit is the mole [mol]. Amount of substance (sometimes abbreviated as 'amount' or 'chemical amount') is proportional to the number NX of specified elementary entities X, and the universal proportionality constant is the reciprocal value of the Avogadro constant (SI),
nX = NX·NA-1 nX contained in a system can change due to internal and external transformations, dnX = dinX + denX In the absence of nuclear reactions, the amount of any atom is conserved, e.g., for carbon dinC = 0. This is different for chemical substances or ionic species which are produced or consumed during the advancement of a reaction r, | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ampere | A | The ampere, symbol A, is the SI unit of electric current. It is defined by taking the fixed numerical value of the elementary charge e to be 1.602 176 634 × 10−19 when expressed in the unit C, which is equal to A s, where the second is defined in terms of ΔνCs. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Avogadro constant | NA [x·mol-1] | {Quote} The Avogadro constant NA is a proportionality constant between the quantity amount of substance (with unit mole) and the quantity for counting entities ... One mole contains exactly 6.022 140 76 × 1023 elementary entities. This number is the fixed numerical value of the Avogadro constant, NA, when expressed in the unit mol−1 and is called the Avogadro number {End of Quote: Bureau International des Poids et Mesures 2019 The International System of Units (SI)}. Thus the Avogadro constant NA has the SI unit 'per mole' [mol-1], but more strictly the unit for counting entities per amount is 'units per mole' [x·mol-1] (compare elementary charge). Therefore, NA is 'count per amount' with units 'counting units per mole'. The Avogadro constant times elementary charge is the Faraday constant. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Barometric pressure | pb [Pa] | Barometric pressure, pb, is an important variable measured for calibration of oxygen sensors in solutions equilibrated with air. The atm-standard pressure (1 atm = 760 mmHg = 101.325 kPa) has been replaced by the SI standard pressure of 100 kPa. The partial pressure of oxygen, pO2, in air is a function of barometric pressure, which changes with altitude and locally with weather conditions. The partial oxygen pressure declines by 12 % to 14 % per 1,000 m up to 6,000 m altitude, and by 15 % to 17 % per 1,000 m between 6,000 and 9,000 m altitude. The O2k-Barometric Pressure Transducer is built into the Oroboros O2k as a basis for accurate air calibrations in high-resolution respirometry. For highest-level accuracy of calculation of oxygen pressure, it is recommended to compare at regular intervals the barometric pressure recording provided by the O2k with a calibrated barometric pressure recording at an identical time point and identical altitude. The concept of gas pressure or barometric pressure can be related to the generalized concept of isomorphic pressure. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Base quantities and count | Template:Base quantities and count | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Body mass | m [kg]; M [kg·x-1] | The body mass M is the mass (kilogram [kg]) of an individual (object) [x] and is expressed in units [kg/x]. Whereas the body weight changes as a function of gravitational force (you are weightless at zero gravity; your floating weight in water is different from your weight in air), your mass is independent of gravitational force, and it is the same in air and water. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boltzmann constant | k [J·x-1·K-1] | The Boltzmann constant k has the SI unit [J·K-1] (IUPAC), but more strictly the units for energy per particles per temperature is [J·x-1·K-1].
k = f·e-1, the electrochemical constant f times the elementary charge e. k = R·NA-1, the gas constant R divided by the Avogadro constant NA. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bound energy | B [J] | The bound energy change in a closed system is that part of the total energy change that is always bound to an exchange of heat,
dB = dU - dA [Eq. 1] ∆B = ∆H - ∆G [Eq. 2] The free energy change (Helmoltz or Gibbs; dA or dG) is the total energy change (total inner energy or enthalpy, dU or dH) of a system minus the bound energy change. Therefore, if a process occurs at equilibrium, when dG = 0 (at constant gas pressure), then dH = dB, and at deW = 0 (dH = deQ + deW; see energy) we obtain the definition of the bound energy as the heat change taking place in an equilibrium process (eq), dB = T∙dS = deQeq [Eq. 3] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Calorespirometric ratio | CR ratio [kJ/mol] | The calorimetric/respirometric or calorespirometric ratio (CR ratio) is the ratio of calorimetrically and respirometrically measured heat and oxygen flux, determinded by calorespirometry. The experimental CR ratio is compared with the theoretically derived oxycaloric equivalent, and agreement in the range of -450 to -480 kJ/mol O2 indicates a balanced aerobic energy budget (Gnaiger and Staudigl 1987). In the transition from aerobic to anaerobic metabolism, there is a limiting pO2, plim, below which CR ratios become more exothermic since anaerobic energy flux is switched on. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Candela | cd | The candela, symbol cd, is the SI unit of luminous intensity in a given direction. It is defined by taking the fixed numerical value of the luminous efficacy of monochromatic radiation of frequency 540 × 1012 Hz, Kcd, to be 683 when expressed in the unit lm W−1. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Canonical ensemble | A canonical ensemble is the group of compartments enclosed in an isolated system H, with a smaller compartment A1 in thermal equilibrium with a larger compartment A2 which is the heat reservoir at temperature T. When A1 is large in the canonical sense, if its state can be described in terms of macroscopic thermodynamic quantities of V, T, and p merging with the state described as a probability distribution. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell ergometry | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Charge | Qel [C] | Charge Qel is the quantity of electricity expressed in the SI unit coulomb [C]. QelX [C] indicates the charge carried by the quantity of a specified ion X. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Charge number | zX | The charge number of an ion X or electrochemical reaction with unit stoichiometric number of X is the particle charge [C·x-1] divided by the elementary charge [C·x-1]. The particle charge QNX is the charge per count of ions X or per ion X transferred in the reaction as defined in the reaction equation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chemical potential | µB [J/mol] | The chemical potential of a substance B, µB [J/mol], is the partial derivative of Gibbs energy, G [J], per amount of B, nB [mol], at constant temperature, pressure, and composition other than that of B,
µB = (∂G/∂nB)T,p,nj≠B The chemical potential of a solute in solution is the sum of the standard chemical potential under defined standard conditions and a concentration (activity)-dependent term, µB = µB° + RT ln(aB)The standard state for the solute is refered to ideal behaviour at standard concentration, c° = 1 mol/L, exhibiting infinitely diluted solution behaviour [1]. µB° equals the standard molar Gibbs energy of formation, ΔfGB° [kJ·mol-1]. The formation process of B is the transformation of the pure constituent elements to one mole of substance B, with all substances in their standard state (the most stable form of the element at 100 kPa (1 bar) at the specified temperature) [2]. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Closed system | A closed system is a system with boundaries that allow external exchange of energy (heat and work), but do not allow exchange of matter. A limiting case is light and electrons which cross the system boundary when work is exchanged in the form of light or electric energy. If the surroundings are maintained at constant temperature, and heat exchange is rapid to prevent the generation of thermal gradients, then the closed system is isothermal. A frequently considered case are closed isothermal systems at constant pressure (and constant volume with aqueous solutions). Changes of closed systems can be partitioned according to internal and external sources. Closed systems may be homogenous (well mixed and isothermal), continuous with gradients, or discontinuous with compartments (heterogenous). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Concentration | c [mol·L-1]; C [x·L-1] | Concentration [mol·L-1] is a volume-specific quantity for diluted samples s. In a concentration, the sample is expressed in a variety of formats: count, amount, charge, mass, energy. In solution chemistry, amount concentration is amount of substance nB per volume V of the solution, cB = [B] = nB·V-1 [mol·dm-3] = [mol·L-1]. The standard concentration, c°, is defined as 1 mol·L-1 = 1 M. Count concentration CX = NX·V-1 [x·L-1] is the concentration of the number NX of elementary entities X, for which the less appropriate term 'number concentration' is used by IUPAC. If the sample is expressed as volume Vs (e.g., VO2), then the 'volume-concentration' of Vs in V is termed 'volume fraction', Φs = Vs·V-1 (e.g., volume fraction of O2 in dry air, ΦO2) = 0.20946). Density is the mass concentration in a volume VS of pure sample S. A change of concentration, dcX, in isolated or closed systems at constant volume is due to internal transformations (advancement per volume) only. In closed compressible systems (with a gas phase), the concentration of the gas changes, when pressure-volume work is performed on the system. In open systems, a change of concentration can additionally be due to external flow across the system boundaries. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Count | NX [x] | 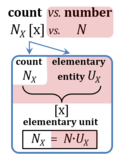 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density | ρ, C, D | Density, mass density ρ = m·V-1 [kg·m-3], is mass m divided by volume V. Surface density ρA = m·A-1 [kg·m-2] (SI). For a pure sample S, the mass density ρS = mS·VS-1 [kg·m-3] is the mass m of pure sample S per volume VS of the pure sample. With density ρ thus defined, the 'amount density' of substance B is ρB = nB·VB-1 [mol·m-3]. This is not a commonly used expression, but the inverse is defined as the molar volume of a pure substance (IUPAC), Vm,B = VB·nB-1 [m3·mol-1]. The pure sample is a pure gas, pure liquid or pure solid of a defined elementary entity. The amount concentration, cB = nB·V-1 [mol·m-3] is the amount nB of substance B divided by the volume V of the mixture (IUPAC), and this is not called an 'amount density'. The term 'amount density' is reserved for an amount of substance per volume VS of the pure substance. This explicit distinction between 'density' related to the volume of the sample and 'concentration' related to the total volume of the mixture is very helpful to avoid confusion. Further clarification is required in cases, when the mass density ρs of the sample in the mixture differs from the mass density ρS of the pure sample before mixing. Think of a sample S of pure ethanol with a volume of 1 L at 25 °C, which is mixed with a volume of 1 L of pure water at 25 °C: after the temperature of the mixture has equilibrated to 25 °C, the total volume of the mixture is less than 2 L, such that the volume VS of 1 L pure ethanol has diminished to a smaller volume Vs of ethanol in the mixture; the density of ethanol in the mixture is higher than the density of pure ethanol (this is incomplete additivity). The volume Vs of sample s in a mixture is by definition smaller than the total volume V of the mixture. Sample volume VS and system volume V are identical, but this applies only to the case of a pure sample. Concentration is related to samples s per total volume V of the mixture, whereas density is related to samples S or s per volume VS = V or Vs < V, respectively (BEC 2020.1). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Dimension | Dimensions are defined in the SI {Quote}: Physical quantities can be organized in a system of dimensions, where the system used is decided by convention. Each of the seven base quantities used in the SI is regarded as having its own dimension. .. All other quantities, with the exception of counts, are derived quantities, which may be written in terms of base quantities according to the equations of physics. The dimensions of the derived quantities are written as products of powers of the dimensions of the base quantities using the equations that relate the derived quantities to the base quantities. There are quantities Q for which the defining equation is such that all of the dimensional exponents in the equation for the dimension of Q are zero. This is true in particular for any quantity that is defined as the ratio of two quantities of the same kind. .. There are also some quantities that cannot be described in terms of the seven base quantities of the SI, but have the nature of a count. Examples are a number of molecules, a number of cellular or biomolecular entities (for example copies of a particular nucleic acid sequence), or degeneracy in quantum mechanics. Counting quantities are also quantities with the associated unit one. {end of Quote: p 136, Bureau International des Poids et Mesures 2019 The International System of Units (SI)} | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discontinuous system | In a discontinuous system, gradients in continuous systems across the length, l, of the diffusion path [m], are replaced by differences across compartmental boundaries of zero thickness, and the local concentration is replaced by the free activity, α [mol·dm-3]. The length of the diffusion path may not be constant along all diffusion pathways, spacial direction varies (e.g., in a spherical particle surrounded by a semipermeable membrane), and information on the diffusion paths may even be not known in a discontinuous system. In this case (e.g., in most treatments of the protonmotive force) the diffusion path is moved from the (ergodynamic) isomorphic force term to the (kinetic) mobility term. The synonym of a discontinuous system is compartmental or discretized system. In the first part of the definition of discontinuous systems, three compartments are considered: (1) the source compartment A, (2) the sink compartment B, and (3) the internal barrier compartment with thickness l. In a two-compartmental description, a system boundary is defined of zero thickness, such that the barrier comparment (e.g., a semipermeable membrane) is either part of the system (internal) or part of the environment (external). Similarly, the intermediary steps in a chemical reaction may be explicitely considered in an ergodnamic multi-comparment system; alternatively, the kinetic analysis of all intermediary steps may be collectively considered in the catalytic reaction mobility, reducing the measurement to a two-compartmental analysis of the substrate and product compartments. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| E | e, E | » Energy, Exergy E
» elementary charge e = 1.602 176 634∙10-19 C∙x-1 » Euler's number e ~ 2.718 281 828 459 » ET capacity E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| E-L coupling efficiency | jE-L | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electric current | Iel [A = C·s-1]; [mol·s-1]; [x·s-1] | Current or electric flow Iel is the advancement of charge per unit of time, expressed in the SI base unit ampere [C·s-1 = A]. Electrons or ions are the current-carrying motive entities of electric flow. Electrons e- are negatively charged subatomic particles carrying 'negative electricity' with a mass that is about 1/1700 of the smallest particle — the proton — carrying 'positive electricity' (Thompson 1906). Correspondingly the velocity of electrons is much higher than that of protons or any other (larger) ion. Current is the velocity v of paticles times the number of motive charges. Therefore, electron current Ie- is of a different nature from electric current Ielχ carried by all species i of ions Xi (cations and anions) summarized as χ = Σ(zi·Xi). Whereas Ie- is the net translocation of electrons moving forwards and backwards, Ielχ is the net translocation of charges carried by different cations and anions. In contrast, ion current IelX of a specific ion X is the partial translocation of charges carried by net translocation of ion X only. If cation current IelX+ is antagonized entirely by counterion current IelY- as the process of antiport, then the electric current Ielχ is zero. The (net) electric current in a compartmental system is driven by the electric force ΔelFp+ or electric potential difference ΔΨp+, whereas a compensated ion/counterion antiport current is insensitive to the electric potential difference. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electric current density | j [C·m-2] | Electric current density is current divided by area, j=I·A-1 [C·m-2]. Compare: density. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrochemical constant | f [J·C-1·K-1] | The electrochemical constant f has the SI unit for energy per charge per temperature [J·C-1·K-1].
f = k·e-1, the Boltzmann constant k divided by the elementary charge e. f = R·F-1, the gas constant R divided by the Faraday constant F. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elementary charge | e [C·x-1] | The elementary charge or proton charge e has the SI unit coulomb [C], but more strictly coulomb per elementary unit [C·x-1]. -e is the charge per electron. Elementary charge e is the charge per elementary entity H+ with SI unit [C] but canonical SI unit [C·x-1]. When the charge Qel [C] of a number Ne [x] of electrons e is divided by the count Ne, then the particle charge QNX (QNX) charge per elementary entity is obtained, -e = Qel/Ne [C·x-1]. e is also used as an atomic unit. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elementary entity | UX [x] | An elementary entity is an entity of type X, distinguished as a single unit of countable objects (X = molecules, cells, organisms, particles, parties, items) or events (X = beats, collisions, emissions, decays, celestial cycles, instances, occurrences, parties). "An elementary entity may be an atom, a molecule, an ion, an electron, any other particle or specified group of particles" (Bureau International des Poids et Mesures 2019). An elementary entity, therefore, needs to be distinguished from non-countable entities and the general class of entities X. This distinction is emphasized by the term 'elementary' (synonymous with 'elementary entity') with symbol UX and elementary unit [x]. If an object is defined as an assembly of particles (a party of two, a molecule as the assembly of a stoichiometric number of atoms), then the elementary is the assembly but not the assembled particle. A number of defined elementaries UX is a count, NX = N·UX [x], where N is a number, and as such N is dimensionless, and N is a number (stop) and is not 'a number of ..'. Elementaries are added as items to a count. The elementary UX has the dimension U of the count NX. The elementary UX has the same unit [x] as the count NX, or more accurately it gives the count the defining 'counting-unit', which is the 'elementary unit' [x]. From the definition of count as the number (N) of elementaries (U) of entity type X, it follows that count divided by elementary is a pure number, N = NX·UX-1. The unit x of a count can neither be the entity X nor a number. The elementary of type X defines the identity X of the elementary UX with the unit 'elementary unit' with symbol [x]. Since a count NX is the number of elementary entities, the elementary UX is not a count (UX is not identical with N·UX). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elementary unit | x | | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Endergonic | Endergonic transformations or processes can proceed in the forward direction only by coupling to an exergonic process with a driving force more negative than the positive force of the endergonic process. The backward direction of an endergonic process is exergonic. The distinction between endergonic and endothermic processes is at the heart of ergodynamics, emphasising the concept of exergy changes, linked to the performance of work, in contrast to enthalpy changes, linked to heat or thermal processes, the latter expression being terminologically linked to thermodynamics. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Endothermic | An energy transformation is endothermic if the enthalpy change of a closed system is positive when the process takes place in the forward direction and heat is absorbed from the environment under isothermal conditions (∆eQ > 0) without performance of work (∆eW = 0). The same energy transformation is exothermic if it proceeds in the backward direction. Exothermic and endothermic transformations can proceed spontaneously without coupling only, if they are exergonic. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Energy | E; various [J] | Heat and work are forms of energy [1 cal = 4.184 J]. Energy [J] is a fundamental term that is used in physics and physical chemistry with various meanings [1]. These meanings become explicit in the following equations relating to systems at constant volume (dV = 0) or constant gas pressure (dp = 0). Energy is exchanged between a system and the environment across the system boundaries in the form of heat, deQ, total or available work, detW (or detW), and matter, dmatU (or dmatH) [2],
dU = (deQ + detW) + dmatU ; dV = 0 [Eq. 1a] dH = (deQ + deW) + dmatH ; dp = 0 [Eq. 1b] Whereas dU (or dH) describe the internal-energy change (or enthalpy change) of the system, heat and work are external energy changes (subscript e; et: external total; e: external excluding pressure-volume work), and dmatU (or dmatH) are the exchange of matter expressed in internal-energy (or enthaply) equivalents. In closed systems, dmatU = 0 (dmatH = 0). The energy balance equation [Eq. 1] is a form of the First Law of Thermodynamics, which is the law of conservation of internal-energy, stating that energy cannot be generated or destroyed: energy can only be transformed into different forms of work and heat, and transferred in the form of matter. Notably, the term energy is general and vague, since energy may be associated with either the first or second law of thermodynamics. Work is a form of energy exchange [Eq. 1], but can be seen as exergy exchange in conjunction with deG = deW in a closed system [Eq. 3b]. An equally famous energy balance equation considers energy changes of the system only, in the most simple form for isothermal systems (dT = 0): dU = dA + T∙dS = dU + dB [Eq. 2a] dH = dG + T∙dS = dG + dB [Eq. 2b] The internal-energy change, dU (enthalpy change, dH) is the sum of free energy change (Helmholtz energy, dA; or Gibbs energy = exergy change, dG) and bound energy change (bound energy, dB = T∙dS). The bound energy is that part of the energy change that is always bound to an exchange of heat. A third energy balance equation accounts for changes of the system in terms of irreversible internal processes (i) occuring within the system boundaries, and reversible external processes (e) of transfer across the system boundaries (at constant gas pressure), dH = diH + deH [Eq. 3a] dG = diG + deG [Eq. 3b] The energy conservation law of thermodynamics (first law) can be formulated as diH = 0 (at constant gas pressure), whereas the generally negative sign of the dissipated energy, diG ≡ diD ≤ 0, is a formulation of the second law of thermodynamics. Insertion into Eq. 3 yields, dH = deH [Eq. 4a] dG = diD + deW + dmatG [Eq. 4b]When talking about energy transformations, the term energy is used in a general sense without specification of these various forms of energy. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enthalpy | H [J] | Enthalpy, H [J], can under conditions of constant gas pressure neither be destroyed nor created (first law of thermodynamics: diH/dt = 0). The distinction between enthalpy and internal-energy of a system is due to external pressure-volume work carried out reversibly at constant gas pressure. The enthalpy change of the system, dH, at constant pressure, is the internal-energy change, dU, minus reversible pressure-volume work,
dH = dU - dVW Pressure-volume work, dVW, at constant pressure, is the gas pressure, p [Pa = J·m-3], times change of volume, dV [m3], dVW = -p·dV [J] The available work, deW, is distinguished from external total work, detW, [1] deW = detW - dVW The change of enthalpy of a system is due to internal and external changes, dH = diH + deH Since diH = 0 (first law of thermodynamics), the dH is balanced by exchange of heat, work, and matter, dH = (deQ + deW) + dmatH ; dp = 0 The exchange of matter is expressed in enthalpy equivalents with respect to a reference state (formation, f, or combustion, c). The value of dH in an open system, therefore, depends on the arbitrary choice of the reference state. In contrast, the terms in parentheses are the sum of all (total, t) partial energy transformations, dtH = (deQ + deW) A partial enthalpy change of transformation, dtrH, is distinguished from the total enthalpy change of all transformations, dtH, and from the enthalpy change of the system, dH. In a closed system, dH = dtH. The enthalpy change of transformation is the sum of the Gibbs energy (free energy) change of transformation, dtrG, and the bound energy change of transformation at constant temperature and pressure, dtrB = T·dS, dtrH = dtrG + dtrB | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Entity | X | An entity of type X is something that can measured as an extensive quantity or counted as an elementary entity. The term entity with symbol X, therefore, has a general meaning, including but not limited to elementary entities UX. The distinction can be emphasized by using the term entity-type X, to avoid confusion of an entity X with the more restricted definition of elementary entity UX as a single countable object or event. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Equality | = | Physicochemical equality (symbol =) indicates in an equation not only numerical equivalence (symbol ≡), but an identity of the full meaning. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Equivalence | ≡ | Numerical equivalence (symbol ≡) indicates that two quantities are numerically equal, even if the full meaning may be different. For instance: 1 ≡ 1·1 and 1 ≡ 1/1. In contrast to ≡, the symbol = indicates physicochemical equality. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ergodynamic efficiency | ε | The ergodynamic efficiency, ε (compare thermodynamic efficiency), is a power ratio between the output power and the (negative) input power of an energetically coupled process. Since power [W] is the product of a flow and the conjugated thermodynamic force, the ergodynamic efficiency is the product of an output/input flow ratio and the corresponding force ratio. The efficiency is 0.0 in a fully uncoupled system (zero output flow) or at level flow (zero output force). The maximum efficiency of 1.0 can be reached only in a fully (mechanistically) coupled system at the limit of zero flow at ergodynamic equilibrium. The ergodynamic efficiency of coupling between ATP production (DT phosphorylation) and oxygen consumption is the flux ratio of DT phosphorylation flux and oxygen flux (P»/O2 ratio) multiplied by the corresponding force ratio. Compare with the OXPHOS-coupling efficiency. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ergodynamics | The mission of ergodynamics is the revelation of relations of general validity. "Thermodynamics deals with relationships between properties of systems at equilibrium and with differences in properties between various equilibrium states. It has nothing to do with time. Even so, it is one of the most powerful tools of physical chemistry" [1]. Ergodynamics is the theory of exergy changes (from the Greek word 'erg' which means work). Ergodynamics includes the fundamental aspects of thermodynamics ('heat') and the thermodynamics of irreversible processes (TIP; nonequilibrium thermodynamics), and thus links thermodynamics to kinetics. In its most general scope, ergodynamics is the science of energy transformations. Classical thermodynamics includes open systems, yet as a main focus it describes closed systems. This is reflected in a nomenclature that is not easily applicable to the more general case of open systems [2]. At present, IUPAC recommendations [3] fall short of providing adequate guidelines for describing energy transformations in open systems. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exergonic | Exergonic transformations or processes can spontaneously proceed in the forward direction, entailing the irreversible loss of the potential to performe work (erg) with the implication of a positive internal entropy production. Ergodynamic equilibrium is obtained when an exergonic (partial) process is compensated by a coupled endergonic (partial) process, such that the Gibbs energy change of the total transformation is zero. Final thermodynamic equilibrium is reached when all exergonic processes are exhausted and all forces are zero. The backward direction of an exergonic process is endergonic. The distinction between exergonic and exothermic processes is at the heart of ergodynamics, emphasising the concept of exergy changes, linked to the performance of work, in contrast to enthalpy changes, linked to heat or thermal processes, the latter expression being terminologically linked to thermodynamics. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exergy | E; various [J] | Exergy includes external and internal work. Exergy as the external work is defined in the First Law of thermodynamics as a specific form of energy. Exergy as the dissipated Gibbs or Helmholtz energy is the irreversibly dissipated (internal) loss of the potential of performing work as defined in the Second Law of Thermodynamics.
Changes of exergy dG plus bound energy yield the enthalpy change: dH = dG + T∙dS = dG + dB | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exothermic | An energy transformation is exothermic if the enthalpy change of a closed system is negative when the process takes place in the forward direction and heat is lost to the environment under isothermal conditions (∆eQ < 0) without performance of work (∆eW = 0). The same energy transformation is endothermic if it proceeds in the backward direction. Exothermic and endothermic transformations can proceed spontaneously without coupling only, if they are exergonic. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Extensive quantity | Extensive quantities pertain to a total system, e.g. oxygen flow. An extensive quantity increases proportional with system size. The magnitude of an extensive quantity is completely additive for non-interacting subsystems, such as mass or flow expressed per defined system. The magnitude of these quantities depends on the extent or size of the system (Cohen et al 2008). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| External flow | Ie [MU·s-1] | External flows across the system boundaries are formally reversible. Their irreversible facet is accounted for internally as transformations in a heterogenous system (internal flows, Ii). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Faraday constant | F [C/mol] | The Faraday constant F links the electric charge [C] to amount [mol], and thus relates the electrical format e [C] to the molar format n [mol]. The Farady constant, F = e·NA = 96 485.33 C/mol, is the product of elementary charge, e = 1.602176634∙10-19 C/x, and the Avogadro constant, NA = 6.02214076∙1023 x/mol. The dimensionless unit [x] is not explicitely considered by IUPAC. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Flow | I [MU∙s-1] | In an isomorphic analysis, any form of flow, I is the advancement of a process per unit of time, expressed in a specific motive unit [MU∙s-1], e.g., ampere for electric flow or current [A≡C∙s-1], watt for heat flow [W≡J∙s-1], and for chemical flow the unit is [mol∙s-1]. Flow is an extensive quantity. The corresponding isomorphic forces are the partial exergy (Gibbs energy) changes per advancement [J∙MU-1], expressed in volt for electric force [V≡J∙C-1], dimensionless for thermal force, and for chemical force the unit is [J∙mol-1], which deserves a specific acronym ([Jol]) comparable to volt. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Flux | J | Flux, J, is a specific quantity. Flux is flow, I [MU·s-1 per system] (an extensive quantity), divided by system size. Flux (e.g., oxygen flux) may be volume-specific (flow per volume [MU·s-1·L-1]), mass-specific (flow per mass [MU·s-1·kg-1]), or marker-specific (e.g. flow per mtEU). The motive unit [MU] of chemical flow or flux is the advancement of reaction [mol] in the chemical format. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Force | F; dmFX; ΔtrFX [J·MU-1] | Force is an intensive quantity. The product of force times advancement is the work (exergy) expended in a process or transformation. Force times flow is power [W].
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Format |  Converstion between different motive formats and corresponding motive units (Gnaiger 2020 BEC MitoPathways) Different formats can be chosen to express physicochemical quantities (motive entities or transformants) in corresponding motive units [MU]. Fundamental formats for electrochemical transformations are:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Free activity | αX [MU·m-3] | Free activity αX [MU·m-3] is pressure divided by isomorphic force. In the chemical amount format, αX is expressed in units of concentration of X [mol·L-1]. αX is the local concentration in a concentration gradient. If the concentration gradient is collapsed to a boundary of zero thickness in a compartmental system, αX reflects the singularity in the transition between the two phases or compartments. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gas constant | R [J·mol-1·K-1] | The gas constant, R = 8.314462618 J·mol-1·K-1, has the SI unit for energy per amount per temperature. R is primarily known from the ideal gas equation, pV = nRT or p = cRT. Therefore, RT is the ratio of pressure p and concentration c.
R = f·F, the electrochemical constant f times the Faraday constant F. R = k·NA, the Boltzmann constant k times the Avogadro constant NA. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gibbs energy | G [J] | Gibbs energy G [J] is exergy which cannot be created internally (subscript i), but in contrast to internal-energy (diU/dt = 0) is not conserved but is dissipated (diG/dt < 0) in irreversible energy transformations at constant temperature and (barometric) pressure, T,p. Exergy is available as work in reversible energy transformations (100 % efficiency), and can be partially conserved when the exergonic transformation is coupled to an endergonic transformation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat | Q, Qth [J] | Heat is a form of energy [J]. The relationship between heat and work provides the foundation of thermodynamics, which describes transformations from an initial to a final state of a system. In energy transformations heat may pass through the boundary of the system, at an external heat flow of deQ/dt. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Holode | Small entetic units are counted into the reference system on a balance opposite to the experimental system with the large sample, which in balance contains as many abstract units as the count of entetic units in the reference system. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hydrogen ion | H+ | The terms hydrogen ion H+ and proton, p or p+, are used synonymously in chemistry. A hydrogen ion is a positively charged molecule. In particle physics, however, a proton is a submolecular and subatomic particle with a positive electric charge. The H+ ion has no electrons and is a bare charge with only about 1/64 000 of the radius of a hydrogen atom. Free H+ is extremely reactive, with an extremely short lifetime in aqueous solutions. There H+ forms the hydronium ion H3O+, which in turn is further solvated by water molecules in clusters such as H5O2+ and H9O4+. The transfer of H+ in an acid–base reaction is referred to as proton transfer. The acid is the H+ donor and the base is the H+ acceptor. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hydron | H+ | Hydron is the general name for the cation H+ used without regard to the nuclear mass of the hydrogen entity (H is the hydro group), either for H in its natural abundance or without distinction between the isotopes. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Iconic symbols | Iconic symbols are used in ergodynamics to indicate more explicitely — compared to standard SI or IUPAC symbols — the quantity represented and some boundary conditions. This is particularly the case in normalized quantities (ratios of quantities). Iconic (or canonical) symbols help to clarify the meaning, are based on SI and IUPAC symbols as far as possible, and may be translated into more commonly used, practical symbols. Several ambiguities in SI and IUPAC symbols are eliminated by the systematic structure of iconic symbols, but it may be impossible to avoid all ambiguities, particulary when long (canonical) symbols are abbreviated in a particular context. Clarity is improved always by showing the unit of a quantity together with the symbol of the quantity. Iconic symbols cannot be identical with IUPAC symbols when a different definition is used — this would add to the confusion. For example, the IUPAC symbols nB [mol] and VB [m3] denote amount and volume of B. Consequently, it should be expected, that the symbol QB indicates charge of B [C]. However, the IUPAC symbol QB is used for particle charge per ion B [C·x-1]. This prohibits a consistent definition of QB as a potential iconic symbol for charge carried by a given quantity of ions B with unit [C], instead of particle charge per ion B with unit [C·x-1]. Hence, the conventional ambigous system forces compatible iconic symbols to be more complicated, using QelB [C] and QNB [C·x-1] to distinguish charge of B from charge per elementary B. QnB [C·mol-1] is charge per molar amount of B. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Intensive quantity | Intensive quantities are partial derivatives of an extensive quantity by the advancement, dtrξX, of an energy transformation tr; example: Force. In contrast to extensive quantities which pertain to the entire system and are additive, extensive quantities 'take well defined values at each point of the system' (Prigogine 1967 Interscience) and are non-additive. Intensive and extensive quantities can be easily discriminated by the units, e.g. [J] for the extensive quantity, in contrast to [J·mol-1] for the corresponding intensive quantity. In the general definition of thermodynamics, intensive quantities are not distinguished from specific quantities (Cohen 2008 IUPAC Green Book). Ergodynamics emphasizes the contrast between specific quantities which are extensive quantities normalized for a variable expressing system size (mass, volume of the system, amount of substance in a system) and intensive quantities which are normalized for the motive unit of a transformation (mass exchanged, volume change of the system, amount of substance reacting in a system; Gnaiger 1993 Pure Appl Chem). Intensive and specific quantities are both non-additive, take well defined values at each point of the system, and both corresponding quantities are expressed in identical units, e.g. the intensive quantity Gibbs force of a catabolic reaction (such as oxidation; 0 = -1 Glc - 6 O2 + 6 CO2 + 6 H2O), ΔkGGlc [kJ·mol-1], and the specific quantity Gibbs energy per mole glucose contained in a system, GGlc [kJ·mol-1] (with respect to an arbitrarily defined reference state, such as the reference state of formation or combustion). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Internal flow | Ii [MU·s-1] | Within the system boundaries, irreversible internal flows, Ii,—including chemical reactions and the dissipation of internal gradients of heat and matter—contribute to internal entropy production, diS/dt. In contrast, external flows, Ie, of heat, work, and matter proceed reversibly across the system boundaries (of zero thickness). Flows are expressed in various formats per unit of time, with corresponding motive units [MU], such as chemical [mol], electrical [C], mass [kg]. Flow is an extensive quantity, in contrast to flux as a specific quantity. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Internal-energy | U [J] | Internal-energy, U [J], can neither be destroyed nor created (first law of thermodynamics: diU/dt = 0). Note that internal (subscript i), as opposed to external (subscript e), must be distinguished from "internal-energy", U, which contrasts with "Helmholtz energy", A, as enthalpy, H, contrasts with Gibbs energy, G. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| International System of Units | SI | The International System of Units (SI) is the modern form of the metric system of units for use in all aspects of life, including international trade, manufacturing, security, health and safety, protection of the environment, and in the basic science that underpins all of these. The system of quantities underlying the SI and the equations relating them are based on the present description of nature and are familiar to all scientists, technologists and engineers. The definition of the SI units is established in terms of a set of seven defining constants. The complete system of units can be derived from the fixed values of these defining constants, expressed in the units of the SI. These seven defining constants are the most fundamental feature of the definition of the entire system of units. These particular constants were chosen after having been identified as being the best choice, taking into account the previous definition of the SI, which was based on seven base units, and progress in science (p. 125). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| International Union of Pure and Applied Chemistry, IUPAC | IUPAC | The International Union of Pure and Applied Chemistry (IUPAC) celebrated in 2019 the 100th anniversary, which coincided with the International Year of the Periodic Table of Chemical Elements (IYPT 2019). IUPAC {Quote} notes that marking Mendeleev's achievement will show how the periodic table is central to connecting cultural, economic, and political dimensions of global society “through a common language” {end of Quote} (Horton 2019). 2019 is proclaimed as the International Year of the Periodic Table of Chemical Elements (IYPT 2019). For a common language in mitochondrial physiology and bioenergetics, the IUPAC Green book (Cohen et al 2008) is a most valuable resource, which unfortunately is largely neglected in bioenergetics textbooks. Integration of open systems and non-equilibrium thermodynamic approaches remains a challenge for developing a common language (Gnaiger 1993; BEC 2020.1). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Isolated system | The boundaries of isolated systems are impermeable for all forms of energy and matter. Changes of isolated systems have exclusively internal origins, e.g., internal entropy production, diS/dt, internal formation of chemical species i which is produced in a reaction r, dini/dt = drni/dt. In isolated systems some internal terms are restricted to zero by various conservation laws which rule out the production or destruction of the respective quantity. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Isomorphic | The term isomorphic refers to quantities which have identical or similar form, shape, or structure. In mathematics, an isomorphism defines a one-to-one correspondence between two mathematical sets. In ergodynamics, isomorphic quantities are defined by equations of identical form. If isomorphic quantities are not expressed in identical units, then these quantities are expressed in different formats which can be converted to identical untis. Example: electric force [V=J/C] and chemical force [Jol=J/mol] are ismorphic forces; the electrical format [J/C] can be converted to the chemical format [J/mol] by the Faraday constant. Units not only give meaning to the numerical value of a quantity, but units provide also an abbreviated common language to communicate and compare isomorphic quantities. In irreversible thermodynamics, isomorphic forces are referred to as generalized forces. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Jmax | Jmax | Jmax is the maximum pathway flux (e.g. oxygen flux) obtained at saturating substrate concentration. Jmax is a function of metabolic state. In hyperbolic ADP or oxygen kinetics, Jmax is calculated by extrapolation of the hyperbolic function, with good agreement between the calculated and directly measured fluxes, when substrate levels are >20 times the c50 or p50. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Kelvin | K | The kelvin, symbol K, is the SI unit of thermodynamic temperature. It is defined by taking the fixed numerical value of the Boltzmann constant k to be 1.380 649 × 10−23 when expressed in the unit J x-1 K−1. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Kilogram | kg | The kilogram, symbol kg, is the SI unit of mass. It is defined by taking the fixed numerical value of the Planck constant h to be 6.626 070 15 × 10−34 when expressed in the unit J s, which is equal to kg m2 s−1, where the meter and the second are defined in terms of c and ΔνCs. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L/E coupling-control ratio | L/E | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Length | l [m] | Length l is an SI base quantity with SI base unit meter m. Quantities derived from length are area A [m2] and volume V [m3]. Length is an extensive quantity, increasing additively with the number of objects. The term 'height' h is used for length in cases of vertical position (see height of humans). Length of height per object, LUX [m·x-1] is length per unit-entity UX, in contrast to lentgth of a system, which may contain one or many entities, such as the length of a pipeline assembled from a number NX of individual pipes. Length is a quantity linked to direct sensory, practical experience, as reflected in terms related to length: long/short (height: tall/small). Terms such as 'long/short distance' are then used by analogy in the context of the more abstract quantity time (long/short duration). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Level flow | E | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Linear phenomenological laws | Linear phenomenological laws are at the core of the thermodynamics of irreversible processes TIP, considered to apply near equilibrium but more generally in transport processes (e.g. Fick's law). In TIP, linearity is discussed as the dependence of generalized flows I or fluxes J on generalized forces, J = -L·F, where L is expected to be constant (as a prerequisite for linearity) and must not be a function of the force F (affinity) for Onsager reciprocity to apply. This paradigm is challenged by the ergodynamic concept of fundamentally non-linear isomorphic flux-force relations and is replaced by the generalized isomorphic flux-pressure relations. Flows I [MU·s-1] and forces F [J·MU-1] are conjugated pairs, the product of which yields power, I·F = P [J·s-1 = W]. Flux J is system-size specific flow, such that volume-specific flux times force yields volume-specific power, PV = J·F [W·m-3]. Then vectoral and vectorial transport processes are inherently non-linear flux-force relationships, with L = u·c in continuous transport processes along a gradient (c is the local concentration), or L = u·α (α is the free activity in a discontinuous transport process across a semipermeable membrane) — formally not different from (isomorphic to) scalar chemical reactions. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Metabolic control variable | X | A metabolic control variable X causes the transition between a background state Y (background rate YX) and a reference state Z (reference rate ZX). X may be a stimulator or activator of flux, inducing the step change from background to reference steady state (Y to Z). Alternatively, X may be an inhibitor of flux, absent in the reference state but present in the background state (step change from Z to Y). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Meter | m | The meter, symbol m, is the SI unit of the SI base quantity length l. It is defined by taking the fixed numerical value of the speed of light c in vacuum to be 299 792 458 when expressed in the unit m·s−1, where the second is defined in terms of the caesium frequency ΔνCs. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mitochondrial membrane potential | mtMP, ΔΨp+, ΔelFep+ [V] | The mitochondrial membrane potential difference, mtMP or ΔΨp+ = ΔelFep+, is the electric part of the protonmotive force, Δp = ΔmFeH+.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mole | mol | The mole [mol] is the SI base unit for the amount of substance of a system that contains 6.02214076·1023 specified elementary entities (see Avogadro constant). The elementary entities must be specified and may be atoms, molecules, ions, electrons, other particles, or specified groups of such particles. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Motive entity | Xtr [MU] |  | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Motive unit | MU | The motive unit [MU] is the variable SI unit in which the motive entity (transformant) of a transformation is expressed, which depends on the energy transformation under study and on the chosen format. Fundamental MU for electrochemical transformations are:
For the protonmotive force the motive entity is the proton with charge number z=1. The protonmotive force is expressed in the electrical or molar format with MU J/C=V or J/mol=Jol, respectively. The conjugated flows, I, are expressed in corresponding electrical or molar formats, C/s = A or mol/s, respectively. The charge number, z, has to be considered in the conversion of motive units (compare Table below), if a change not only of units but a transition between the entity elementary charge and an entity with charge number different from unity is involved (e.g., O2 with z=4 in a redox reaction). The ratio of elementary charges per reacting O2 molecule (zO2=4) is multiplied by the elementary charge (e, coulombs per proton), which yields coulombs per O2 [C∙x-1]. This in turn is multiplied with the Avogadro constant, NA (O2 molecules per mole O2 [x∙mol-1]), thus obtaining for zeNA the ratio of elementary charges [C] per amount of O2 [mol-1]. The conversion factor for O2 is 385.94132 C∙mmol-1. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Net P/E control ratio | (P-L)/E | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Net R/E control ratio | (R-L)/E | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Number | N, n | A number N (or n) is a count NX [x] divided by the elementary entity UX [x]. X must represent the same entity in both occurences. The elementary unit [x] cancels in the division by simplification, such that numbers (for example, numbers 8 or 24) are abstracted from the counted entity X. The concept of number is tightly entangled with units, counts and entities. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Numeral | A numeral is the symbol representing a specific number. A numeral is the figure of a number, with different notation types used as a figure (VIII and 8 for Roman and Arabic numerals; 八 and 捌 for practical and financial Chinese). A numeral may consist of one or more characters or digits. 60 and 60.00 are different numerals consisting of two and four digits, respectively, which represent the same number sixty. Sixty is the name of the number 60, with the meaning 'number 60'. N is not a numeral but a symbol representing the entity 'number'. The equation N=60 assignes the numerical value 60 to the entity 'number'. The numeral 60 is a symbol for a pure number that equals 6 times 10 (or 2 times 30; or 1 times 60). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Open system | An open system is a system with boundaries that allow external exchange of energy and matter; the surroundings are merely considered as a source or sink for quantities transferred across the system boundaries (external flows, Iext). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxygen flow | IO2 [mol·s-1] or [mol·s-1·x-1] | Respiratory oxygen flow is the oxygen consumption per total system, which is an extensive quantity. Flow is advancement of a transformation in a system per time [mol·s-1], when 'system' is defined as the experimental system (e.g. an open or closed chamber). Flow is distinguished from the size-specific quantity flux obtained by normalization of flow per volume of the experimental system [mol·s-1·m-3]. An experimental object, e.g. a living cell, may be considered as the 'experimental system'. Then oxygen flow per cell has the unit [mol·s-1·x-1], where [x] is the elementary unit for a count. Oxygen flow or respiration per cell [amol·s-1·x-1] = [pmol·s-1·Mx-1] is normalized for the cell count, distinguished from oxygen flux (e.g. per mg protein or wet mass). These are different forms of normalization of rate. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxygen flux | JO2 | Oxygen flux, JO2, is a specific quantity. Oxygen flux is oxygen flow, IO2 [mol·s-1 per system] (an extensive quantity), divided by system size. Flux may be volume-specific (flow per volume [pmol·s-1·mL-1]), mass-specific (flow per mass [pmol·s-1·mg-1]), or marker-specific (flow per mtEU). Oxygen flux (e.g., per body mass, or per cell volume) is distinguished from oxygen flow (per number of objects, such as cells), IO2 [mol·s-1·x-1]. These are different forms of normalization of rate. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxygen pressure | pO2 [kPa] | Oxygen pressure or partial pressure of oxygen [kPa], related to oxygen concentration in solution by the oxygen solubility, SO2 [µM/kPa]. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxygen solubility | SO2 [µM/kPa] | The oxygen solubility, SO2 [µM/kPa] = [(µmol·L-1)/kPa], expresses the oxygen concentration in solution in equilibrium with the oxygen pressure in a gas phase, as a function of temperature and composition of the solution. The inverse of oxygen solubility is related to the activity of dissolved oxygen. The oxygen solubility in solution, SO2(aq), depends on temperature and the concentrations of solutes in solution, whereas the dissolved oxygen concentration at equilibrium with air, cO2*(aq), depends on SO2(aq), barometric pressure and temperature. SO2(aq) in pure water is 10.56 µM/kPa at 37 °C and 12.56 µM/kPa at 25 °C. At standard barometric pressure (100 kPa), cO2*(aq) is 207.3 µM at 37 °C (19.6 kPa partial oxygen pressure) or 254.7 µM at 25 °C (20.3 kPa partial oxygen pressure). In MiR05 and serum, the corresponding saturation concentrations are lower due to the oxygen solubility factor: 191 and 184 µM at 37 °C or 234 and 227 µM at 25 °C. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P-L control efficiency | jP-L | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P/E control ratio | P/E | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PH | pH | The pH value or pH is the negative of the base 10 logarithm of the activity of protons (hydrogen ions, H+). A pH electrode reports the pH and is sensitive to the activity of H+. In dilute solutions, the hydrogen ion activity is approximately equal to the hydrogen ion concentration. The symbol pH stems from the term potentia hydrogenii. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Particle charge | QNX, QNX | The particle charge QNX (QNX) or charge per elementary entity is the charge QelX [C] carried by ions of type X divided by the count NX [x]. The particle charge per proton is the elementary charge or proton charge e. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pascal | Pa | The pascal [Pa] is the SI unit for pressure. [Pa] = [J·m-3] = [N·m-2] = [m-1·kg·s-2]. The standard pressure is 100 kPa = 1 bar (105 Pa; 1 kPa = 1000 Pa). Prior to 1982 the standard pressure has been defined as 101.325 kPa or 1 standard atmosphere (1 atm = 760 mmHg). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pressure | p, P, Π [Pa] | Pressure is a fundamental quantity expressing energy per volume. The SI unit of pressure is generally pascal [Pa] = [J·m-3]. The term 'stress' (mechanical stress) is used as a synonym for pressure (SI). Pressure is known in physics as mechanical pressure, which is force per area, p = F·A-1 [Pa] = [N·m-2]. In physical chemistry, gas pressure is defined as p = n·V-1·RT, where the concentration is c = n·V-1 [mol·m-3], R is the gas constant, and T is the absolute temperature, and RT is expressed in units of chemical force [J·mol-1]. van't Hoff's osmotic pressure assumes the same form applied to dissolved substances diffusing across a semipermeable membrane, but concentrations should be replaced by activities. The activity of dissolved gases is expressed by the partial pressure, where the solubility can be seen as an activity coefficient. Pressure appears explicitely or implicitely in all chapters of physics and physical chemistry. In contrast to the universal counterparts energy and force, however, the general connections between various isomorphic expressions of pressure remain poorly understood: Pressure is the concentration of the force at the point of action. More generally, pressure is the force times concentration at the interphase of interaction. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Proton | p+, p | The terms proton p and hydrogen ion H+ are used synonymously in chemistry. In particle physics, a proton is a subatomic particle with a positive electric charge. Protons and neutrons are collectively referred to as nucleons. The proton is a bare charge with only about 1/64 000 of the radius of a hydrogen atom, and so the free proton is extremely reactive chemically. Therefore, the free proton has an extremely short lifetime in aqueous solutions where it forms the hydronium ion, H3O+, which in turn is further solvated by water molecules in clusters such as H5O2+ and H9O4+. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Protonmotive force | pmF, ∆mFH+, Δp [J·MU-1] | The protonmotive force ∆mFH+ is known as Δp in Peter Mitchell’s chemiosmotic theory [1], which establishes the link between electric and chemical components of energy transformation and coupling in oxidative phosphorylation. The unifying concept of the pmF ranks among the most fundamental theories in biology. As such, it provides the framework for developing a consistent theory and nomenclature for mitochondrial physiology and bioenergetics. The protonmotive force is not a vector force as defined in physics. This conflict is resolved by the generalized formulation of isomorphic, compartmental forces, ∆trF, in energy (exergy) transformations [2]. Protonmotive means that there is a potential for the movement of protons, and force is a measure of the potential for motion.
The pmF is generated in oxidative phosphorylation by oxidation of reduced fuel substrates and reduction of O2 to H2O, driving the coupled proton translocation from the mt-matrix space across the mitochondrial inner membrane (mtIM) through the proton pumps of the electron transfer pathway (ETS), which are known as respiratory Complexes CI, CIII and CIV. ∆mFH+ consists of two partial isomorphic forces: (1) The chemical part, ∆dFH+, relates to the diffusion (d) of uncharged particles and contains the chemical potential difference§ in H+, ∆µH+, which is proportional to the pH difference, ∆pH. (2) The electric part, ∆elFp+ (corresponding numerically to ∆Ψ)§, is the electric potential difference§, which is not specific for H+ and can, therefore, be measured by the distribution of any permeable cation equilibrating between the negative (matrix) and positive (external) compartment. Motion is relative and not absolute (Principle of Galilean Relativity); likewise there is no absolute potential, but isomorphic forces are stoichiometric potential differences§. The total motive force (motive = electric + chemical) is distinguished from the partial components by subscript ‘m’, ∆mFH+. Reading this symbol by starting with the proton, it can be seen as pmF, or the subscript m (motive) can be remembered by the name of Mitchell, ∆mFH+ = ∆dFH+ + ∆elFp+ With classical symbols, this equation contains the Faraday constant, F, multiplied implicitly by the charge number of the proton (zH+ = 1), and has the form [1] ∆p = ∆µH+∙F-1 + ∆ΨA partial electric force of 0.2 V in the electrical format, ∆elFeH+a, is 19 kJ∙mol-1 H+a in the molar format, ∆elFnp+a. For 1 unit of ∆pH, the partial chemical force changes by -5.9 kJ∙mol-1 in the molar format, ∆dFnH+a, and by 0.06 V in the electrical format, ∆dFeH+a. Considering a driving force of -470 kJ∙mol-1 O2 for oxidation, the thermodynamic limit of the H+a/O2 ratio is reached at a value of 470/19 = 24, compared to the mechanistic stoichiometry of 20 for the N-pathway with three coupling sites. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Protonmotive pressure | pmP, ∆mΠH+ [kPa] | The protonmotive pressure, ∆mΠH+ or pmP [kPa], is an extension of Peter Mitchell’s concept of the protonmotive force pmF, based on Fick’s law of diffusion and Einstein’s diffusion equation, accounting for osmotic pressure (corresponding to the diffusion term in the pmF) and electric pressure (the electric term or membrane potential in the pmF). The linearity of the generalized flow-pressure relationship explains the non-ohmic flow-force dependence in the proton leak rate as a function of membrane potential.
The total motive pressure (motive = electric + chemical) is distinguished from the partial components by subscript ‘m’, ∆mΠH+, ∆mΠH+ = ∆dΠH+ + ∆elΠp+ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Quantities, symbols, and units | In the context of quantities, symbols, and units, a code is required to convert terms defining physicochemical quantities into symbols (encoding) and to decode symbols as used in equations, text, and figures. Then symbols and abbreviations gain meaning. Simple symbols — such as Q or N — are used with different meanings depending on context (think of Q for heat and Q for electric charge; or N for number of cells and N for number of O2 molecules). The context provides the code. When the context is extended, the symbols have to be expanded too, including more detail to avoid confusion (Qth versus Qel; Nce versus NO2). Then symbols may appear too complicated, loosing the function of sending their message quickly. There is no single best way to design the right symbol or to replace meaningful symbols (Qel) by ambiguous abbreviations (Q) — all depends on context. We need to use the adequate medium (words, symbols, and abbreviations; equations, text, and figures; videos and slide presentations) and provide the code to achieve communication. The medium is the message, the message is the meaning — from Marshall McLuhan to Hofstadter. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Quantity | Q | A quantity is the attribute of a phenomenon, body or substance that may be distinguished qualitatively and determined quantitatively. A dimensional quantity is a number (variable, parameter, or constant) connected to its dimension, which is different from 1. {Quote} The value of a quantity is generally expressed as the product of a number and a unit. The unit is simply a particular example of the quantity concerned which is used as a reference, and the number is the ratio of the value of the quantity to the unit. {end of Quote: Bureau International des Poids et Mesures 2019 The International System of Units (SI), p. 127)}. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RT | RT versus RT | RT indicates room temperature or 25 °C. RT is the gas constant R [kJ/mol] multiplied by absolute temperature T [K]. This is the motive force quantum in the amount format (Gnaiger 2020 BEC MitoPathways). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SI base units | Template:Keywords: SI base units 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SI prefixes | There are 20 SI prefixes defined to represent multiples and submiltiples of SI units. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| STPD | STPD | At standard temperature and pressure dry (STPD: 0 °C = 273.15 K and 1 atm = 101.325 kPa = 760 mmHg), the molar volume of an ideal gas, Vm, and Vm,O2 is 22.414 and 22.392 L∙mol-1, respectively. Rounded to three decimal places, both values yield the conversion factor of 0.744 from units used in spiroergometry (VO2max [mL O2·min-1]) to SI units [µmol O2·s-1]. For comparison at normal temperature and pressure dry (NTPD: 20 °C), Vm,O2 is 24.038 L∙mol-1. Note that the SI standard pressure is 100 kPa, which corresponds to the standard molar volume of an ideal gas of 22.711 L∙mol-1 and 22.689 L∙mol-1 for O2. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Scalar | A scalar is a pysicochemical quantity that is fully described by its magnitude. A potential difference, differences of concentration or pressure are scalars, whereas a potential gradient is a vector. Similarly, the protonmotive force and metabolic oxygen flux are scalars, whereas the fundamental forces of physics and velocity are vectors. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Second | s | The second, symbol s, is the SI unit of time. It is defined by taking the fixed numerical value of the caesium frequency ∆νCs, the unperturbed ground-state hyperfine transition frequency of the caesium 133 atom, to be 9 192 631 770 when expressed in the unit Hz, which is equal to s−1. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sides | There are many sides of the term 'side' in our language system. Inside and outside are the sides that are separated by the system boundaries of an experimental system. + and - are the two sides of numbers separated by 0. Pages in books have opposite sides or front sides versus backsides. Many fundamental terms have opposite sides of the meaning, thus spanning the entire message in the space between their apparently contrasting sides, and transforming the paradox as a perspective into the unified whole, the full, the complete. On the other side, such fundamental terms are fully understood only after realization of the opposite sides of their meaning — treasures discovered in the etymological origins of the word. It makes sense to open all our senses to comprehend the bright side and the dark side of things. Whereas the student sais "I see a black sheep", Zen decides "You see, that one side of the sheep is black". This is the message to consider both sides before choosing sides, besides overcoming a one-sided point of view. Don't rock side-to-side, but get immersed deeply inside things to see the upsides and downsides of every thing or anything, and more so of nothing. Inside is the insight, for insiders and outsiders of the feedback loop of an Ouroboros. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solubility | SG | The solubility of a gas, SG, is defined as concentration divided by partial pressure, SG = cG·pG-1. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solutions | A solution is {Quote}: A liquid or solid phase containing more than one substance, when for convenience one (or more) substance, which is called the solvent, is treated differently from the other substances, which are called solutes. When, as is often but not necessarily the case, the sum of the mole fractions of solutes is small compared with unity, the solution is called a dilute solution. A superscript attached to the ∞ symbol for a property of a solution denotes the property in the limit of infinite dilution {end of Quote: IUPAC Gold Book}. » MiPNet article | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Specific quantity | Specific quantities are obtained when the extensive quantity is divided by system size, in contrast to intensive quantities. The adjective specific before the name of an extensive quantity is often used to mean divided by mass (Cohen et al 2008). A mass-specific quantity (e.g., mass-specific flux is flow divided by mass of the system) is independent of the extent of non-interacting homogenous subsystems. If mass-specific oxygen flux is constant and independent of system size (expressed as mass), then there is no interaction between the subsystems. The well-established scaling law in respiratory physiology reveals a strong interaction of oxygen consumption and body mass by the fact that mass-specific basal metabolic rate (oxygen flux) does not increase proportionally and linearly with body mass. Maximum mass-specific oxygen flux, VO2max, is less mass-dependent across a large range of body mass of different mammalian species (Weibel and Hoppeler 2005). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed | v [m·s-1] | Speed, v [m·s-1], is the distance, s [m], covered by a particle per unit time, irrespective of geometrical direction in space. Therefore, speed is not a vector, in contrast to velocity. v = ds/dt [m·s-1] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Static head | L | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stoichiometric number | νX | The sign of the stoichiometric number νX is determined by the nonspatial direction of the transformation (positive for products, negative for substrates), and the magnitude of νX is determined by the stoichiometric form. For instance, νA=-1 in the reaction 0 = -1 A + 2 B (-1 glucose converted to +2 lactate), but νA=-1/6 in the reaction 0 = -1/6 A - 1 B + 1 C (-1/6 glucose and -1 O2 converted to +1 H2CO3). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Subscripts in physical chemistry | Subscripts in physical chemistry are used to differentiate symbols of different quantities. While these subscripts need to be short to be readable, they have to be distinct and well defined. Several subscripts relate to fundamental terms and concepts, summarized in a list below. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| System | The term system has a variety of meanings and dictionary definitions in different contexts, e.g., the International System of Units (SI), MKSA system, data management system, biological or mechanical system, redox system, Electron transfer system, loosely or completely coupled system, instrumental system. In thermodynamics and ergodynamics, the system is considered as an experimental system (experimental chamber), separated from the environment as an isolated, adiabatic, closed, or open system. {Quote } The internal domain of any system is separated from the external domain (the surroundings) by a boundary. In theory, energy transformations outside the system can be ignored when describing the system. The surroundings are merely considered as a source or sink for quantities transferred across the system boundary. According to the transfer properties of the boundary, three types of thermodynamic systems are distinguished. (1) The boundaries of isolated systems are impermeable for all forms of energy and matter. Isolated systems do not interact with the surroundings. Strictly, therefore, internal changes of isolated systems cannot be observed from outside since any observation requires interaction. (2) The boundaries of closed systems are permeable for heat and work, but impermeable for matter. A limiting case is electrons which cross the system boundary when work is exchanged in the form of electric energy [added: and light]. The volume of a closed system may be variable. (3) The boundaries of open systems allow for the transfer of heat, work and matter. Changes of isolated systems have exclusively internal origins, whereas changes of closed and open systems can be partitioned according to internal and external sources. Production and destruction of a quantity within the system are internal changes, whereas changes of heat, work and matter due to transfer across the system boundaries are labelled extenal. (External) transfer is thus contrasted with (internal) production or destruction. {end of Quote: Gnaiger 1993 Pure Appl Chem} A system may be treated as a black box. In the analysis of continuous or discontinuous systems, however, information is implied on the internal structure of the system. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit | UX [x]; uQ | | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vector | A vector is a pysicochemical quantity with magnitude and spatial direction of a gradient. Symbols for vectors are written in bold face. For example, velocity, v, and the fundamental forces of physics, F, are vectors. An infinitesimal area is a vector, dA, perpendicular to the plane. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Velocity | v [m·s-1] | Velocity, v [m·s-1], is the speed in a defined spatial direction, and as such velocity is a vector. Velocity is the advancement in distance per unit time, v ≡ dz ∙ dt-1 [m·s-1] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Volume | V [m3]; 1 m3 = 1000 L | Volume V is a derived quantity based on the SI base quantity length [m] and is expressed in terms of SI base units in the derived unit cubic meter [m3]. The liter [L = dm3] is a conventional unit of volume for concentration and is used for most solution chemical kinetics. The volume V contained in a system (experimental chamber) is separated from the environment by the system boundaries; this is called the volume of the system, and described in practical language as big/small (derived from length, height) or voluminous. Systems are defined at constant volume or constant pressure. For a pure sample S, the volume VS of the pure sample equals the volume V of the system, VS = V. For sample s in a mixture, the ratio Vs·V-1 is the nondimensional volume fraction Φs of sample s. Quantities divided by volume are concentrations of sample s in a mixture, such as count concentration CX = NX·V-1 [x·L-1], and amount of substance concentration CB = nB·V-1 [mol·L-1]. Mass concentration is density ρs = ms·V-1 [kg·L-1]. In closed compressible systems (with a gas phase), the concentration of the gas increases, when pressure-volume work is performed on the system. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Work | deW [J] | Work [J] is a specific form of energy in the First Law of thermodynamics, and a specific form of exergy in the Second Law of thermodynamics, performed by a closed or open system on its surroundings (the environment). This is the definition of external work, which is zero in isolated systems. The term exergy includes external and internal work. Mechanical work is force [N] times path length [m]. The internal-energy change of a closed system, dU, is due to external exchange (e) of work and heat, and external total work (et, including pressure-volume work) is the internal-energy change minus heat, detW = dU - deQ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≡ | ≡ | The symbol ≡ indicates (numerical) equivalence, in contrast to = as the symbol for (physicochemical) equality. |
- »Bioblast links: Energy and exergy - >>>>>>> - Click on [Expand] or [Collapse] - >>>>>>>
- Units
- Joule [J]; 1 J = 1 N·m = 1 V·C; 1 cal = 4.184 J
- Units
- Fundamental relationships
- » Energy
- » Exergy
- » Extensive quantity
- Fundamental relationships
- Contrast
- » Force
- » Pressure
- » Intensive quantity
- Contrast
- Forms of energy
- » Internal-energy dU
- » Enthalpy dH
- » Heat deQ
- » Bound energy dB
- Forms of energy
- Forms of exergy
- » Helmholtz energy dA
- » Gibbs energy dG
- » Work deW
- » Dissipated energy diD
- Forms of exergy
- Bioblast links: Force and membrane potential - >>>>>>> - Click on [Expand] or [Collapse] - >>>>>>>
- Fundamental relationships
- mt-Membrane potential and protonmotive force
- O2k-Potentiometry
- » O2k-Catalogue: O2k-TPP+ ISE-Module
- » O2k-Manual: MiPNet15.03 O2k-MultiSensor-ISE
- » TPP - O2k-Procedures: Tetraphenylphosphonium
- » Specifications: MiPNet15.08 TPP electrode
- » Poster
- » Unspecific binding of TPP+
- » TPP+ inhibitory effect
- O2k-Potentiometry
- O2k-Fluorometry
- » O2k-Catalogue: O2k-FluoRespirometer
- » O2k-Manual: MiPNet22.11 O2k-FluoRespirometer manual
- » Safranin - O2k-Procedures: MiPNet20.13 Safranin mt-membranepotential / Safranin
- » TMRM - O2k-Procedures: TMRM
- O2k-Fluorometry
- O2k-Publications
- Bioblast links: SI base units - >>>>>>> - Click on [Expand] or [Collapse] - >>>>>>>
- Entity, count, and number, and SI base quantities / SI base units
Quantity name Symbol Unit name Symbol Comment elementary UX elementary unit [x] UX, UB; [x] not in SI count NX elementary unit [x] NX, NB; [x] not in SI number N - dimensionless = NX·UX-1 amount of substance nB mole [mol] nX, nB electric current I ampere [A] A = C·s-1 time t second [s] length l meter [m] SI: metre mass m kilogram [kg] thermodynamic temperature T kelvin [K] luminous intensity IV candela [cd]
- Fundamental relationships
- » Avogadro constant NA
- » Boltzmann constant k
- » elementary charge e
- » Faraday constant F
- » gas constant R
- » electrochemical constant f
- Fundamental relationships
- SI and related concepts
- Bioblast links: System - >>>>>>> - Click on [Expand] or [Collapse] - >>>>>>>
- Bioblast links: Concentration and pressure - >>>>>>> - Click on [Expand] or [Collapse] - >>>>>>>
- Concentration
- » Volume
- » Activity
- » Concentration
- » Density
- » Mole
- » Molar mass
- Concentration
- Pressure
- Solubility = concentration/pressure
- General
- » Boltzmann constant
- » Energy
- » Force
- » Gas constant
- » Work
- General
- Related keyword lists